")
教程:AMBER计算离子液体径向分布函数(RDFs)、自扩散系数...手册介绍
- 1 创建初始结构
- 1.1 用xleap绘制分子
- 1.2 创建pdb文件
- 1.3 重复步骤
- 2 Antechamber
- 2.1 产生乙腈的mol2和frcmod文件
- 2.2 硼原子的问题
- 2.3 在xLEaP中输入
- 3 Parmchk
- 4 Packmol
- 5 使用tLEaP生成Amber prmtop文件
- 6 用Sander进行最小化
- 7 运行分子动力学模拟
- 8 用ptraj成像
- 9 自扩散系数
- 10 结论
- 11 参考文献
用xleap绘制分子
- 2022-03-16 17:49:36
- 青
- 4580
- 最后编辑:默尼化工科技(上海)有限公司 于 2022-03-16 18:19:35
We will use
xleap to draw the molecules and to generate pdb files. First type “xleap” at the command prompt to bring up a window like this one:
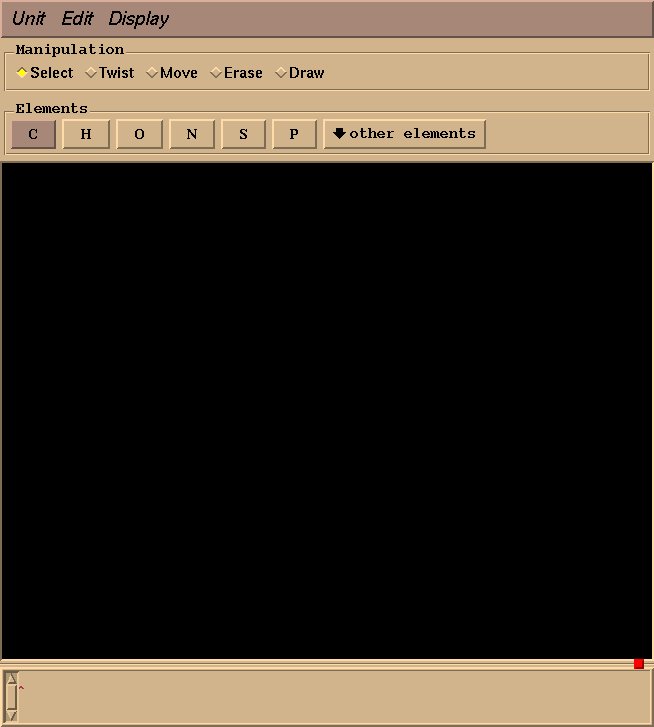
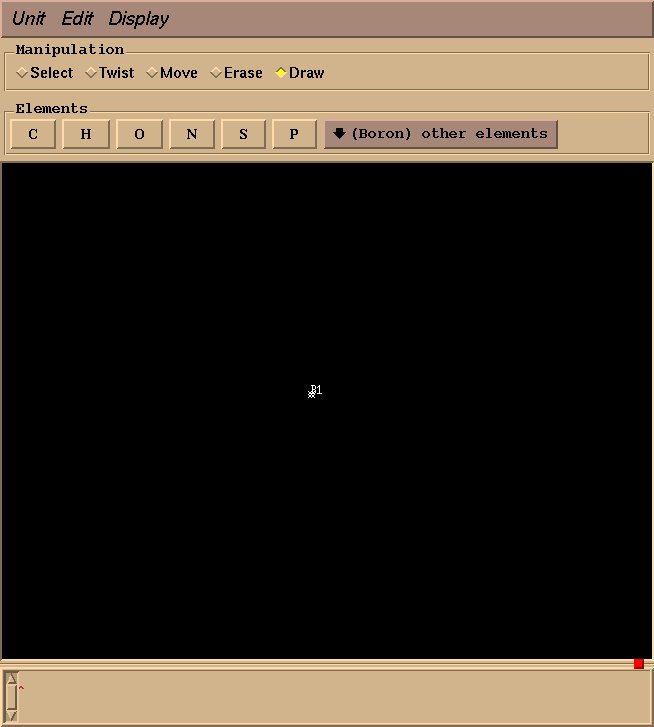
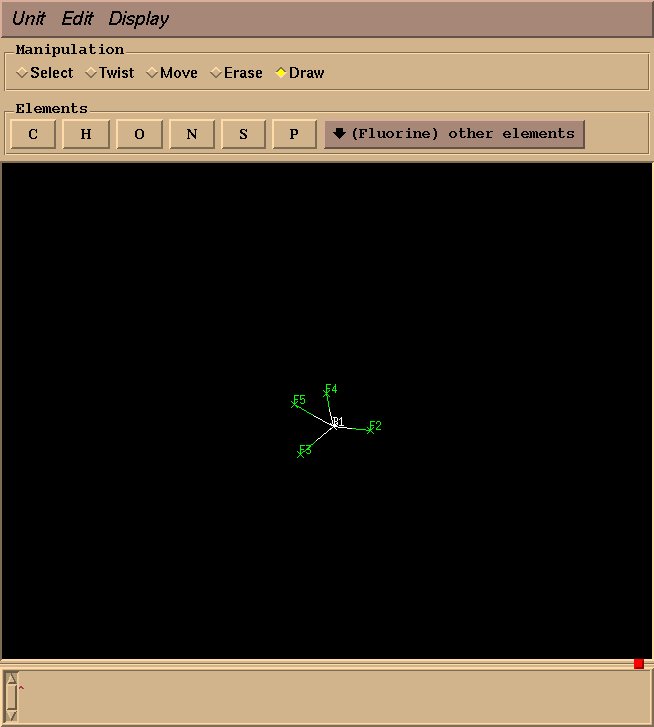
This will create an “BF4” object, and a graphical interface will open pop up. Drawing a molecule is fairly intuitive: select “draw” in the “Manipulation” box, select an appropriate element, click to draw an atom, and drag to draw a bond. For BF4, select “Boron” from “other elements,” and left click to draw a single boron atom.
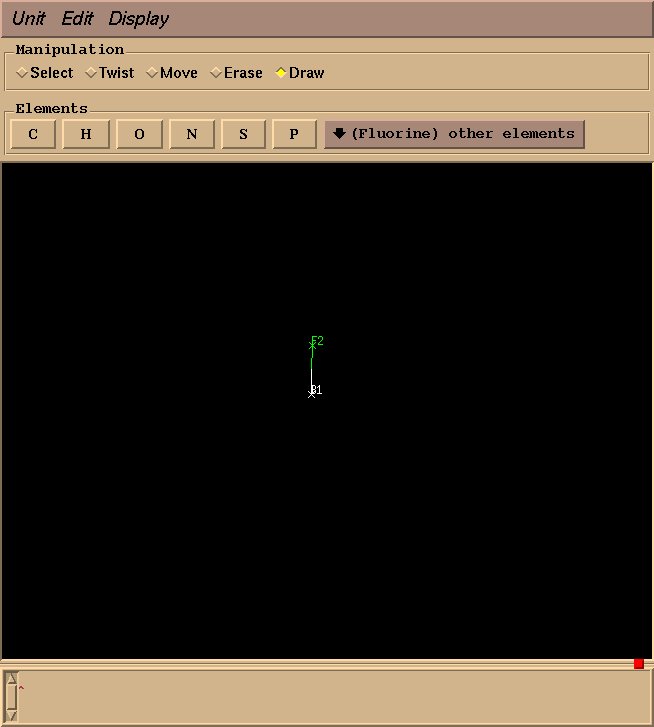
Next, select “Fluorine” from “other elements,” and drag your mouse from the boron atom to the desired fluorine position.
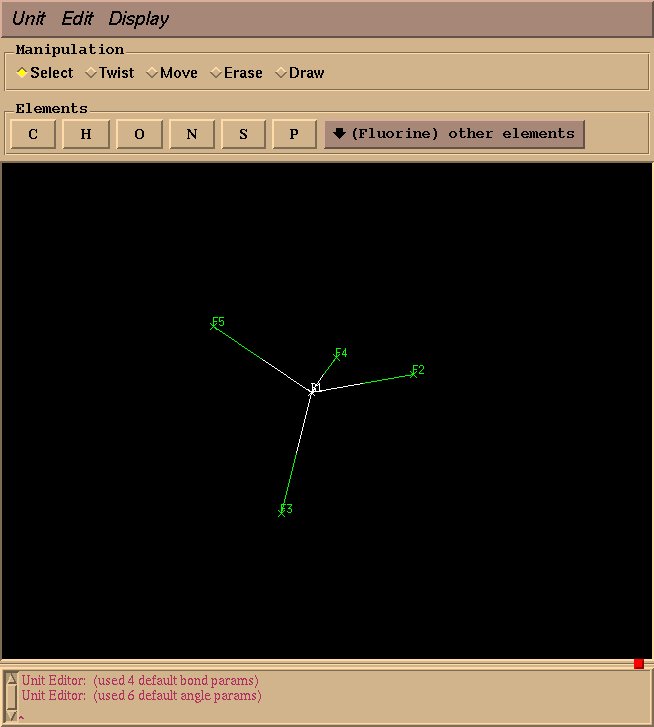
Draw three more fluorine atoms in the same manner, roughly in a tetrahedral geometry. The geometry need not be perfect at this stage–rough estimations will suffice.
Note: Hold control and move the mouse to rotate the view. Hold control and the right mouse while moving the mouse upwards to zoom in or downwards to zoom out. If you click an atom and select it, hold shift and click anywhere in the drawing space to deselect.
发表评论
ICP标识
底部分享
产品
会议
学习
关于 ILPlatform
“离子液体(ILs)产学研”平台(ILPlatform),用于打造出ILs内容自由分享集散中心,基于该中心将关联到与ILs有关的各个资源,为ILs从业人员以及想了解ILs的人们提供便捷的信息获取与分享渠道,未来实现多样化终端部署,让信息传递更加及时有效...